시스템생물학의 합성생물학 적용을 위한 표준화
이충훈 박사, 김지현 교수
연세대학교 시스템생물학과
1. 시스템생물학과 합성생물학
1990년대 중반부터 생물학계에서는 생명 현상을 단편적인 현상이 아닌 하나의 시스템 수준에서 분석하고 이해하려는 시도가 시작되었고, 이러한 움직임은 시스템생물학이라는 이름으로 널리 알려지게 되었다. 세포 주기에 대한 수학적 모델 수립에서 촉발된 시스템생물학은 (Tyson, 2001, Novak, 1993, Chen, 2004), 이내 그 활용도를 크게 인정받아 의약 및 다양한 생명공학 분야에 적용되기 시작하였고 (Kitano 2002a, Kitano 2002b, Kitano 2000, Ideker 2001), 최근에는 항암 치료제의 임상 실험에도 수학적 모델을 이용한 시스템생물학적 접근방법이 사용되고 있다 (Schoeberl, 2010).
생명체를 시스템 수준에서 분석하고 이해할 수 있게 해 준 시스템생물학의 발전은, 다양한 생명체 내의 기능 모듈들을 화학적으로 합성할 수 있게 해준 기술의 발달을 통해, 시스템 레벨에서의 이해를 시스템 레벨에서의 설계 및 조작으로 응용하고자 하는 합성생물학의 발전으로 자연스럽게 이어졌다.
대용량으로 얻어지는 유전체 및 각종 오믹스 데이터들을 시스템 레벨에서 분석함으로써, 생명체 안의 여러 인자들이 서로 어떻게 반응하면서 네트워크를 형성하고 있는지를 유전체, 전사체, 단백질체, 대사체 등 다양한 레벨에서 확인할 수 있게 되었고, 이들을 통해 네트워크 내의 어떠한 부분을 건드려야 원하는 효과를 얻을 수 있는지, 네트워크 내부의 어떠한 모듈이 네트워크 안에서 어떠한 기능을 하는지에 대한 보다 정확한 정보를 얻을 수 있게 되었을 뿐만 아니라, 새로운 유용한 기능의 모듈에 대한 정보를 확보할 수 있게 되었다.
또한 재구성된 네트워크를 바탕으로 시뮬레이션을 수행함으로써, 새로운 모듈을 외부에서 넣어주었을 때 전체 네트워크가 이에 대해 어떻게 반응하는지, 예상치 못한 상호작용으로 인한 부작용은 없는지, 다양한 환경 변화에 대해서도 새롭게 구성된 시스템이 안정적으로 작동할 수 있는지 등을 인실리코로 미리 테스트해볼 수 있게 되었다. 다시 말해서, 시스템생물학의 발전을 통해 합성생물학은 생물학적 시스템의 설계를 위한 강력한 도구를 확보할 수 있게 되었다 (그림 1).
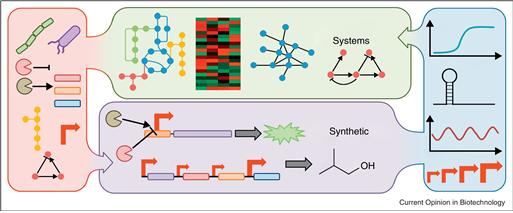
그림 1. 시스템생물학과 합성생물학 사이의 시너지 효과 (Lanza, 2012)
2. 오믹스 정보 메타데이터 및 데이터 저장 방식 표준화
생명체 내부의 변화를 측정하기 위한 다양한 기술적 발전들은 이제 생명체 내부의 작은 모듈들의 변화를 시스템 전체적으로 관찰하고 기록할 수 있게끔 해주고 있다. 특히 Next Generation Sequencing (NGS) 기술의 발전은 유전체의 염기서열 뿐만 아니라 전사체에 대한 대용량 정보 확보 또한 싸고 빠르게 달성할 수 있게 해주고 있다. 기술의 발달이 데이터의 확보를 용이하게 해 주었다면, 문제는 이러한 데이터를 어떻게 분석할 것인가 하는 부분이다. 수십 기가에 달하는 분석 정보들이 수 시간 안에 쏟아져 나오지만, 이러한 데이터들 속에서 제대로 된 분석 결과를 얻기란 쉬운 일이 아니다.
데이터 분석의 첫 번째 단계는 어떠한 데이터를 보관할 것인가 하는 것에서 시작된다. 단순히 오믹스 정보를 축적한다고 해서 그 데이터가 의미를 가지는 것은 아니다. 모든 오믹스 데이터들에 있어서 가장 중요한 정보는 그 자신보다는 그 자신이 얻어지게 된 환경적 요인에 대한 메타 데이터다. 어떠한 균주에 대한 데이터인지, 어떠한 조건에서 추출한 데이터인지를 정확하게 알고 있어야만 이들을 비교한 결과가 생물학적 의미를 부여받을 수 있다. 데이터를 얻은 상황에 대한 정확한 설명이 존재하지 않을 경우, 이를 이용한 분석은 잘못된 결과로 오도될 수 있는 여지가 존재하기 때문이다.
Minimum Information About a Microarray Experiment (MIAME) (Brazma, 2001), Minimum Information About a Proteomic Experiment (MIAPE) (Taylor, 2007, Martens, 2008) 그리고 Minimum Information for Biological and Biomedical Investigation (MIBBI) project (Taylor, 2008) 같은 표준들은 어떠한 메타 데이터들이 오믹스 데이터의 분석에 있어 필수적인 요소들인지를 제시하고자 하는 노력의 결과물들이다. 이들은 각각 전사체, 단백질체, 그리고 그 외의 다양한 생물학적 실험들에 있어서 어떠한 정보가 상호간의 비교를 위해 중요한 역할을 하는지에 대한 가이드라인을 제시하고 있다. 이러한 표준화된 비교 기준이 있을 때, 다양한 플랫폼과 다양한 조건하에 얻어진 각종 오믹스 데이터들이 오류 없이 비교되어질 수 있다 (그림 2).
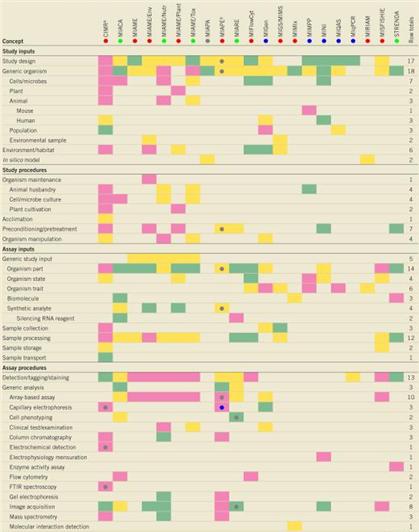
그림 2. MIBBI에 포함된 메타데이터 정보 예시 (Taylor, 2008)
3. 네트워크 재구성 수준 벤치마킹 방법 표준화
다양한 대용량 오믹스 데이터를 얻은 이후 이를 분석할 때 다음으로 문제가 되는 것은, 이들을 이용해 생물학적 네트워크를 재구성하게 될 경우, 과연 얼마나 정확하게 재구성이 되었는지를 판단하기 어렵다는 부분이다. 생물학적 네트워크가 가지고 있는 복잡성은 그 안에 존재하는 구성원들의 수의 증가에 따라 기하급수적으로 증가하게 된다. 때문에 단순한 테스트만으로 이들 네트워크가 얼마나 잘 구성되었는지를 판단하기란 불가능하다. 그렇다고 모든 가능성에 대해 테스트를 수행하는 일도 불가능에 가깝다.
대조군 설계는 얼마나 잘 이루어졌는지, 샘플링 시점은 어떻게 정하였는지, 얼마나 정교하게 반복 실험이 디자인되었는지 하는 부분들은 추후 재구성되는 네트워크의 신뢰도를 크게 좌우하게 된다. 또한, 사용된 통계학적 기법 내지는 수학적 방법론들은 어떠한 것인지, 어디까지 모델이 단순화되어 있는지, 어떠한 가정들이 사용되었는지에 대한 부분 역시 네트워크 재구성에 있어 그 정확도와 신뢰도에 큰 영향을 준다. 지금까지 이러한 부분에 있어서 벤치마킹을 위한 기준의 부재가 하나의 문제로 대두되고 있었다.
Dialogue for Reverse Engineering Assessments and Methods (DREAM) initiative (De Smet, 2010)는 이러한 부분에 있어서 서로 다른 생물학적 데이터 및 네트워크 분석 정보들을 비교할 수 있는 기준을 제시하려는 시도이다. DREAM을 통해 다양한 생물학적 데이터들과 이들을 분석한 다양한 통계학적 분석 방법 내지는 수학적 알고리즘들이 비교되었고, 그 결과 다양한 방법론들이 컨텍스트에 의존적으로 서로 보완적인 기능을 할 수 있으며, 이를 바탕으로 그들 사이에서 공통적으로 나타나는 결과를 종합해 분석함으로써, 높은 퀄리티의 네트워크 재구성이 가능하다는 것이 확인되었다 (그림 3).
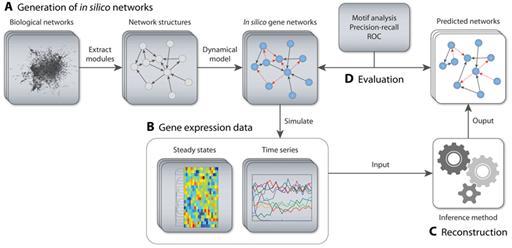
그림 3. 오믹스데이터로부터 네트워크를 재구성하고 벤치마킹하는 것에 대한 예시 (Schaffter, 2011)
4. 생물학적 네트워크 정보 기록 방법 표준화
네트워크의 재구성이 원활하게 이루어졌다 하더라도, 이것이 활용하기 용이한 방식으로 데이터베이스화되어있지 않다면, 이를 활용하기란 여간 어려운 일이 아니다. 네트워크의 구성요소 및 그 안에서의 상호작용을 사람이 혹은 컴퓨터가 읽기 쉽도록 만들어주지 않는다면, 매번 이러한 정보를 컴퓨터가 읽기 쉬운 형태로 재가공하는 일이 필요하다. 역으로 컴퓨터만 읽기 쉬운 형태로 정보가 저장되어 있을 경우, 이를 사람이 보고 활용하기가 쉽지 않다.
따라서 이러한 네트워크에 대한 다양한 정보들이 컴퓨터가 읽기 쉬운 형태로, 혹은 사람이 읽기 쉬운 형태로 각각 저장하기 위한 표준이 존재해야 이러한 정보들이 손쉽게 활용될 수 있을 뿐만 아니라, 이들 사이의 변환을 위한 도구의 개발을 통해서 사람과 컴퓨터 양쪽 모두에서 최소한의 노력을 통해 생물학적 네트워크에 대한 정보를 활용할 수 있게 된다.
이러한 표준화에 대한 노력의 결과물로 Systems Biology Markup Language (SBML) (Hucka, 2003)과 Biological Pathways exchange (BioPAX) (Demir, 2010)의 경우에는 다양한 Bio-molecule들 간의 네트워크를 컴퓨터가 읽기 쉽게 표현하기 위한 표준으로 개발되어 널리 사용되어지고 있는 반면, Systems Biology Graphical Notation (SBGN) (Le Novere, 2009)의 경우는 사람이 읽기 더 편하도록 도식화하여 기록하는 표준으로 개발되어 사용되어지고 있다 (그림 4).
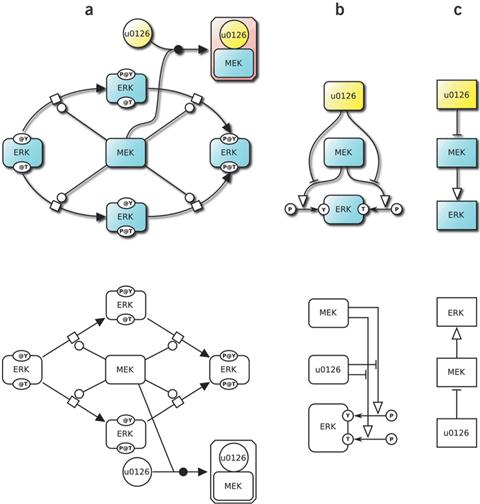
그림 4. SBGN diagram 예시 (Le Novere, 2009)
5. 시뮬레이션 메타데이터 및 결과 데이터 기록 방법 표준화
오믹스 데이터를 얻어내는 동안의 변인들에 대한 메타 데이터, 이들을 이용해 네트워크를 재구성하는 방식에 대한 비교, 네트워크를 기록하고 저장하는 방식들 등에 대한 표준화된 기준들 외에 추가적으로 필요한 것이 이렇게 재구성된 네트워크를 이용해 시뮬레이션을 수행했을 때 이에 대한 정보를 어떻게 저장하고, 시뮬레이션 결과의 비교에 활용할 것인가에 대한 표준화된 기준이다.
Deterministic model인지 stochastic model인지, dynamic model인지 constraint-based model인지, partial differential equation을 사용하는지, ordinary differential equation을 사용하는지와 같은 시뮬레이션에 있어서의 방법론에 따라서 시뮬레이션 결과가 달라질 수 있을 뿐만 아니라, 동일한 방법론을 통해 네트워크의 동작을 시뮬레이션 하더라도, 그 안에 존재하는 다양한 파라미터들의 변화에 따라 시뮬레이션 결과가 크게 달라질 수 있다.
즉, 이러한 시뮬레이션 실험에 대한 사용된 툴이나 파라미터에 대한 메타 데이터들을 어떻게 기록하고 저장할 것인지에 대한 표준화된 기준이 존재해야, 그 결과들을 오류 없이 비교하고 분석하는 것이 가능해진다.
Simulation Experiment Description Markup Language (SED-ML) (Waltemath, 2011)의 경우에는 시뮬레이션에 사용된 모델의 종류, 각종 수학적 파라미터들의 수치들 및 아웃풋의 형식에 대한 정보를 XML 형식으로 담도록 하는 표준으로 개발되었다 (그림 5). Systems Biology Results Markup Language (SBRML) (Dada, 2010)은 SBML을 보완하기 위해 개발된 표준으로, SBML 모델을 이용해 시뮬레이션을 수행한 결과들을 어떻게 저장할 것인지를 지정하고 있다.
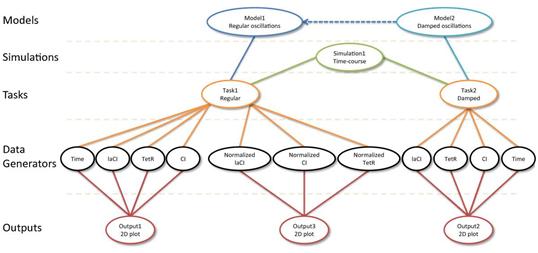
그림 5. SED-ML을 통해 기록되는 시뮬레이션 정보 (Waltemath, 2011)
6. 고찰
다양한 오믹스 데이터의 시스템 수준에서의 분석 및 시뮬레이션을 위한 기술들의 급격한 발전은, 관련 논문들과 데이터들의 기하급수적인 증가라는 결과로 나타나고 있다. 그러나 이들에 대한 제대로 된 표준화가 이루어지지 않을 경우, 광대한 데이터들이 서로 연계되어 분석되지 못하고 사장되기 십상이다. 따라서 오믹스 데이터들은 습득에서부터 저장, 분석 및 시뮬레이션에 이르기까지의 모든 과정들이 표준화된 기준에 따라 그 설정과 결과 데이터들이 함께 저장되고 분석되어야 하며, 이것이 이루어졌을 때 이들을 바탕으로 한 시스템의 설계 및 합성이 성공적으로 수행될 수 있을 것이다.
참고문헌
1. Tyson, J. J., Chen, K. & Novak, B., Network dynamics and cell physiology, 2001, Nature Rev. Mol. Cell Biol., 2, 908-16.
2. Novak, B. & Tyson, J. J., Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos, 1993, J. Cell Sci., 106, 1153–68.
3. Chen, K. C. et al., Integrative analysis of cell cycle control in budding yeast, 2004, Mol. Biol. Cell, 15, 3841–62.
4. Kitano, H., Systems biology: a brief overview, 2002, Science, 295, 1662–4.
5. Kitano, H., Computational systems biology, 2002, Nature, 420, 206–10.
6. Kitano, H., Perspectives on systems biology, 2000, New Generation Computing, 18, 199–216.
7. Ideker, T., Galitski, T. & Hood, L., A new approach to decoding life: systems biology, 2001, Annu. Rev. Genomics Hum. Genet., 2, 343–72.
8. Schoeberl, B. et al., An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation, 2010, Cancer Res., 70, 2485–94.
9. Lanza, A. M., Crook, N. C. & Alper, H. S., Innovation at the intersection of synthetic and systems bioloogy, 2012, Curr. Opin. Biotech., 23, 712-7.
10. Brazma, A. et al., M., Minimum information about a microarray experiment (MIAME) — toward standards for microarray data, 2001, Nature Genet., 29, 365–71.
11. Taylor, C. F. et al., The minimum information about a proteomics experiment (MIAPE), 2007, Nature Biotech., 25, 887–93.
12. Martens, L., Palazzi, L. M. & Hermjakob, H., Data standards and controlled vocabularies for proteomics, 2008, Methods Mol. Biol., 484, 279–86.
13. Taylor, C. F. et al., Promoting coherent minimum reporting guidelines for biological and biomedical investigations: the MIBBI project, 2008, Nature Biotech., 26, 889–96.
14. De Smet, R. & Marchal, K., Advantages and limitations of current network inference methods, 2010, Nature Rev. Microbiol., 8, 717–729.
15. Schaffter, T., Marbach, D. & Floreano, D., GeneNetWeaver: in silico benchmark generation and performance profiling of network inference methods, 2011, Bioinformatics, 27, 2263-70.
16. Hucka, M. et al., The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models, 2003, Bioinformatics, 19, 524–31.
17. Demir, E. et al., The BioPAX community standard for pathway data sharing, 2010, Nature Biotech., 28, 935–42.
18. Le Novere, N. et al., The Systems Biology Graphical Notation, 2009, Nature Biotech., 27, 735–41.
19. Waltemath, E. et al., Reproducible computational biology experiments with SED-ML – the Simulation Experiment Description Markup Language, 2011, BMC Syst. Biol., 5:198.
20. Dada, J. O. et al., SBRML: a markup language for associating systems biology data with models, 2010, Bioinformatics, 26:932-8.







 시스템생물학의 합성생물학 적용을 위한 표준화(연세대 김지현 교수, 이충훈 박사)
시스템생물학의 합성생물학 적용을 위한 표준화(연세대 김지현 교수, 이충훈 박사)